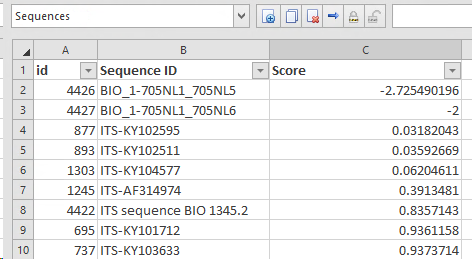
*Make sure there is a D field in the target table (in this case in the sequence table) the field here is called ‘Score’ and will be used to write the results of the curation.
-
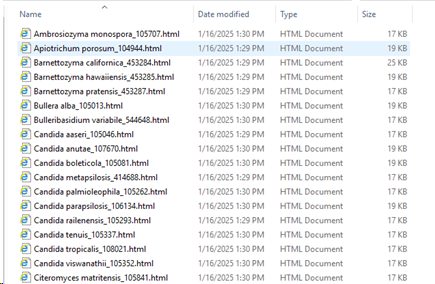
Do a search for the wanted records in the main grid of BioloMICS.
The tool will take all records currently in the grid (all pages takes).
So if a part of the data should be used, then first do a query to get the wanted records.
-
For example, search for all records that have an ITS sequence linked.
-
Go to Other Tools and click on Automated curation.
-
Popup window appears with a short explanation.
-
The 'one field' will be explained in this chapter and is to point out the outsiders at the unit level (sequence level in this case).
-
Select first option: ‘
Select one field only for the automated curation’.
It will use the one selected field to find outliers and bad data points.
-
-
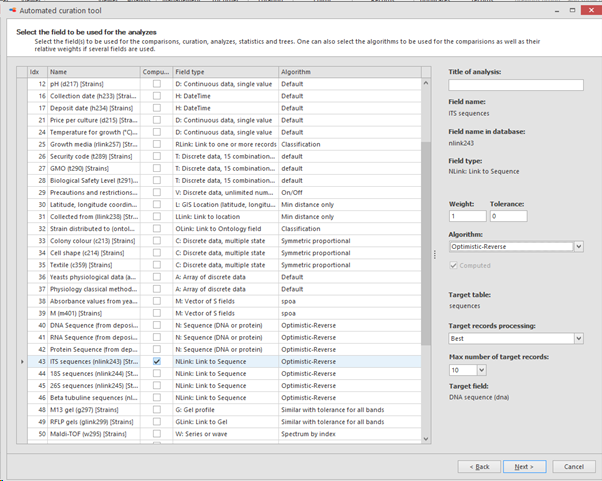
In the next step select the field to be used for the comparisons, curation, analyzes, statistics and trees.
-
On the right, the algorithm can be selected that will be used for the comparisons.
Here we will select the sequence link-field for ITS sequences.
-
-
Then select the options for the analyzes:
Hover the mouse on top of the number to see the details.
-
-
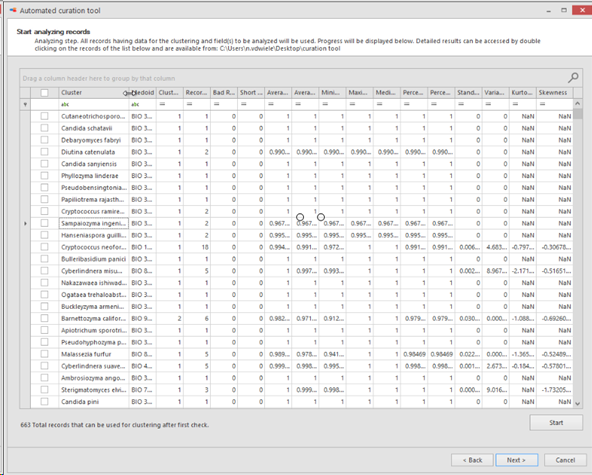
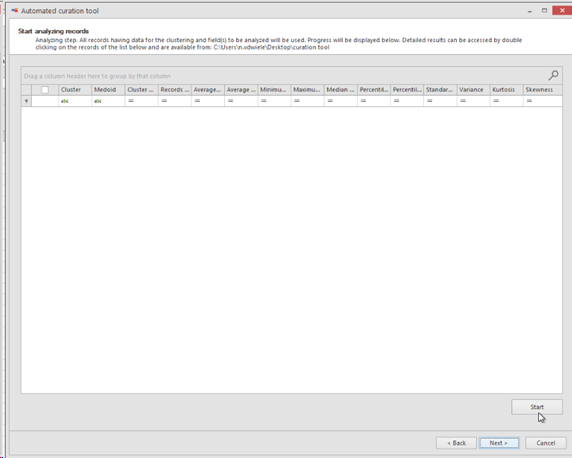
Analyzing step. All records having data for the clustering and field(s) to be analyzed will be used.
Progress will be displayed at the bottom of the popup window.
Detailed results can be accessed by double clicking on the records of the provided list and are available from the provided path place (#13).
-
Now it will do the following:
-
Loading selected records.
-
Checking for each sequence if the length is long enough to be part of the analysis, otherwise it will be rejected and it gets a score of -2.
-
Analyzing cluster
per species name (in this case).
-
All found
species names (in this case) are listed together with extra information in separate columns:
-
Cluster:
Species name (in this case).
-
Medoid: Name of the strain that is indicating the medoid of the given species.
-
Medoid = the one with the smallest distance to all other point in the same species (in this case).
-
Cluster number: Number of cluster(s) found. Note that the bad and short
sequences are excluded.
-
Record number: Number of records found.
-
Bad records: Number of bad records found.
-
Short records: Number of short records found.
-
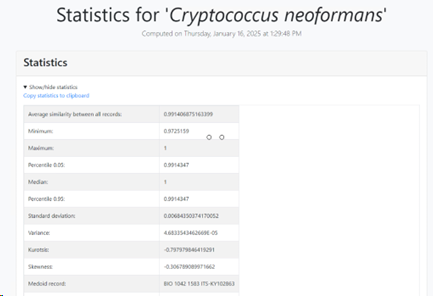
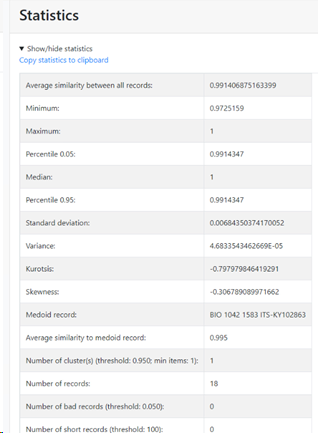
Average similarity to Medoid: Average similarity to medoid record.
-
Average similarity: Average similarity between all records.
-
Minimum similarity: The furthest pair of points within the
species.
-
Maximum similarity: The closest pair of points within the
species.
-
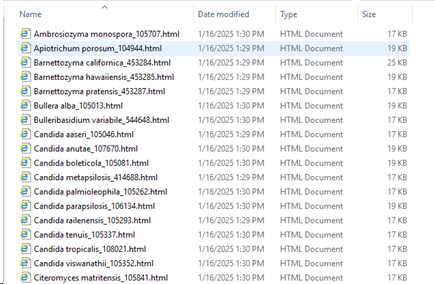
Double click on a given
species name to open the HTML document and to see all the details. Note that when there is only 1 record in the group then the report is not generated.
-
-
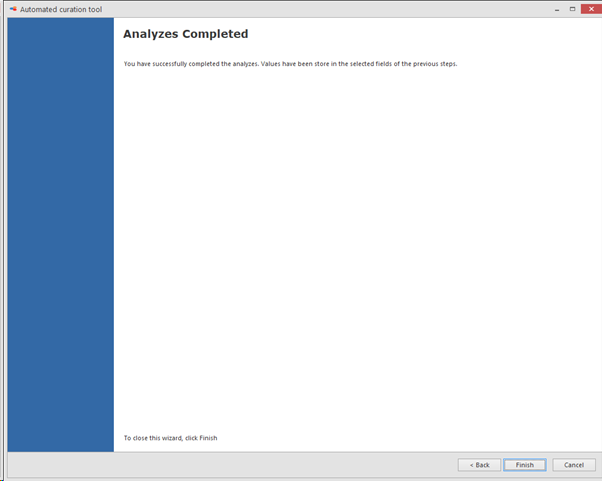
Click next to continue and to complete the analyzes.
-
Completion of the analyzes.
-
This movie shows how to use the Automated curation tool using one field only.
1. Search for wanted records (0:09)
2. Start Automated curation (0:28)
3. Select one field (0:51)
4. Select field to curate (0:59)
5. Set options (1:13)
6. Start analyzing records (3:51)
7. Read results (4:12)
8. Complete wizard (7:23)
9. Check score (7:28)
All needed information is given above this line.
_________________________________________________________________________________________
|
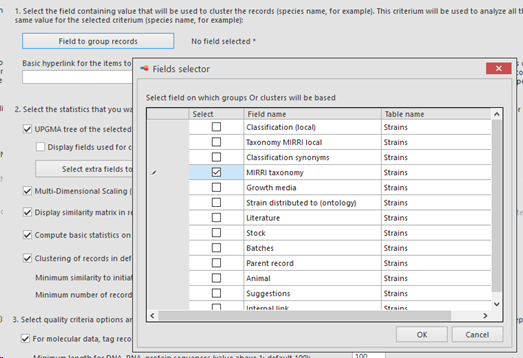
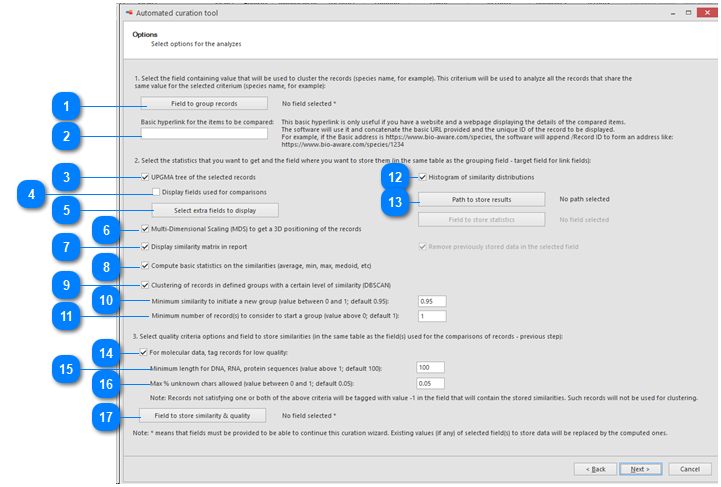
Field to group records: Select the field containing the value that will be used to cluster the records (species name for example).
This criterium will be used to analyze all the records that share the same value for the selected criterium (species name for example).
-
Here we will select MIRRI taxonomy. So this means that the ITS sequences (field was selected in previous step) will be grouped based on the taxon name value (species name) in the MIRRI taxonomy link-field. So all Candida albicans’ together and all Cryptococcus neoformans’ together etc. 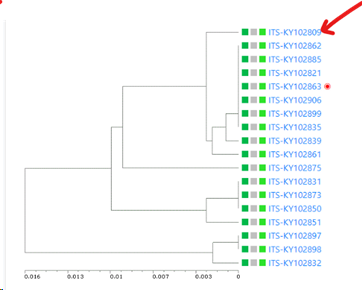
-
Higher level grouping (items will be grouped at genus level instead of species): If checked then the items will be grouped based on the level above, so if the record name contains the species name then in this case the genus will be used for the grouping.
-
For Synlink fields: it takes the value before the first space that is the higher level. And groups based on that first text. -
For Olink fields: the parent is taken.
|
|
|
Basic hyperlink for the items to be compared: This hyperlink is useful when there is a website displaying the details of the compared items.
The link will be presented in the clustering tree in the results.
The software will use the provided basic hyperlink and concatenate it with the unique ID of the record to be displayed.
For example, if the basic address is https://www.bio-aware.com/species, the software will append /Record ID to form and address like: https://www.bio-aware.com/species/1234.
If no basic hyperlink is provided then there won’t be a hyperlink in the clustering tree in the results.
|
|
|
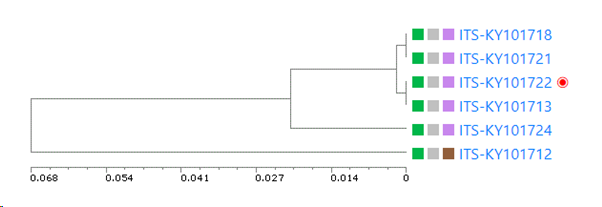
UPGMA tree of the selected records: if this checkbox is checked then there will be a specific section in the results that shows the tree. If unchecked then this section is not shown in the results.
|
|
|
Display fields used for comparisons: Check the checkbox to show the selected field from the previous step (ITS sequences here) in the first column in the tree.
|
|
|
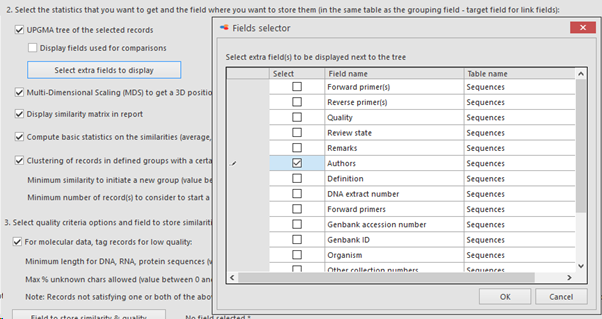
Select extra fields to display: When more field details should be included in the tree, here we added 1 extra field.
The selected field(s) will be shown between the first column (the selected field from point 5, ITS sequence here) and the last column (indicating the cluster the item is in). So if 1 extra field is selected, then 1 extra column is present (see picture below), when 3 extra fields are selected then 3 extra columns are in the between the first and the last column, 
|
|
|
Multi-Dimensional Scaling (MDS) to get a 3D positioning of the records: if this checkbox is checked then there will be a specific section in the results that shows the MDS. If unchecked then this section is not shown in the results.
|
|
|
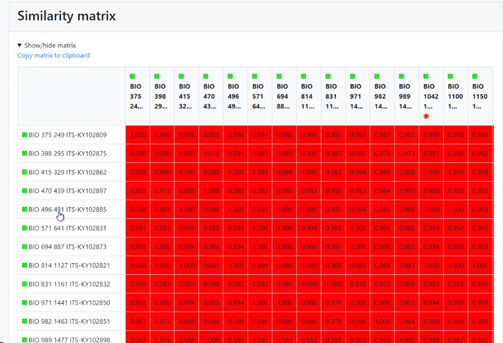
Display similarity matrix in report: if this checkbox is checked then there will be a specific section in the results that shows the similarity matrix between the items in the comparison (sequences in this case). If unchecked then this section is not shown in the results.
|
|
|
Compute basic statistics on the similarities (average, min, max, medoid, etc): if this checkbox is checked then there will be a specific section in the results that shows the basic statistics (on top). If unchecked then this section is not shown in the results.
|
|
|
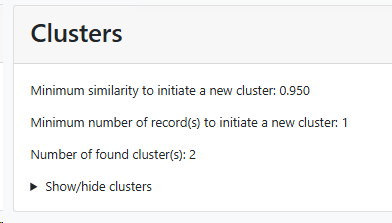
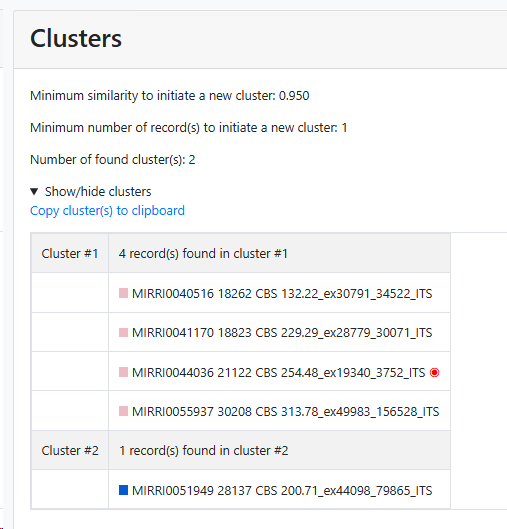
Clustering of records in defined groups with a certain level of similarity (DBSCAN):
When this checkbox the next 2 options (#10 and #11) are enabled.
|
|
|
Only activated when #9 is checked.
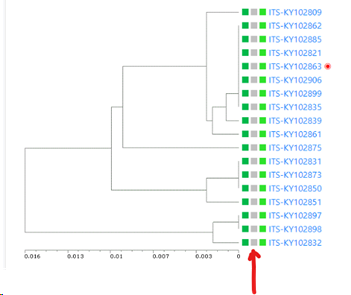
Minimum similarity to initiate a new group (value between 0 and 1; default 0.95): When the default of 0.95 is kept then it means that when a sequence is more than 5% different from the others (any other) then it will be placed in a separate cluster.
Here for example the similarity of the last sequence is 0.068 (6.8%), which is more than the 5% that was the limit, therefore it is placed in a separate cluster.
|
|
|
Minimum number of record(s) to consider to start a group (value above 0, default 1): Enter the number of records that should be at least in a group before it can be called a group.
|
|
|
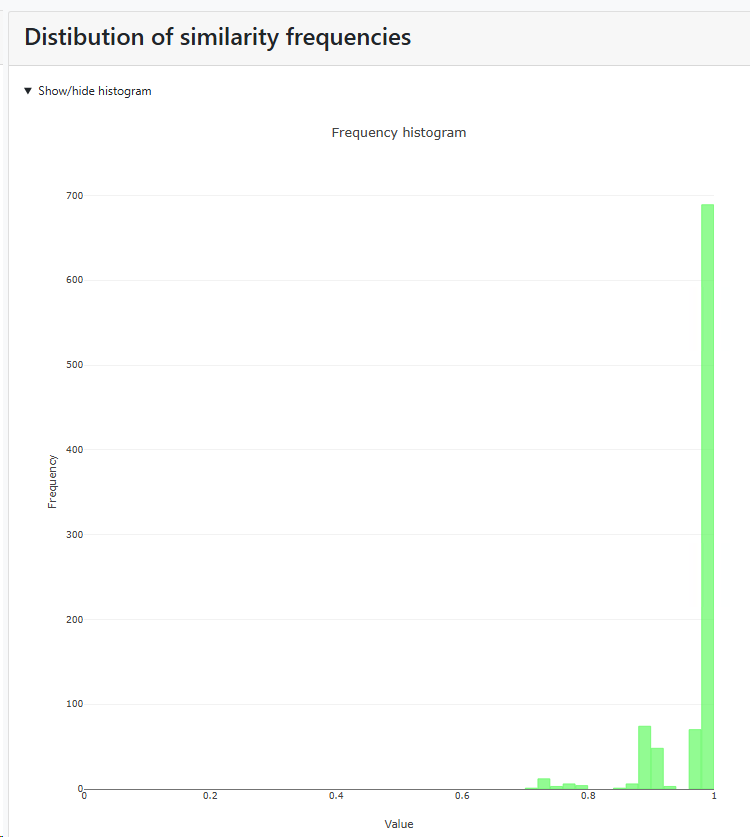
Histogram of similarity distributions: if this checkbox is checked then there will be a specific section in the results that shows the distribution of similarity frequencies. If unchecked then this section is not shown in the results.
|
|
|
Path to store results: The results will be stored in separate HTML docs. One doc per species (in this case). Select the place where to store these documents.
|
|
|
For molecular data, tag records for low quality: When the checkbox is checked then the sequences will be checked based on their length and their percentage ATCG. If a sequence is not fitting the criteria then it will be tagged (noted in the field to be provided in ‘Field to store similarity & quality’), and may therefore be a potential problem that needs to be looked at.
|
|
|
Minimum length for DNA, RNA, protein sequences (value above 1; default 100): the minimum length of the sequence to include it in the analysis. If the sequence is shorter than the provided value then it will be excluded.
-
The scores are explained in the separate Scores section.
|
|
|
Max % unknown chars allowed (value between 0 and 1; default 0.05): The maximum percentage of non-ATCG characters to include it in the analysis. If the percentage of non-ATCG present in the sequence is higher than the provided value then it will be excluded.
|
|
|
Field to store similarity & quality: Select the field that will store the scores per item (sequence). This must be a D-field, in the target table (here the Sequences table).
Note that new scores will overwrite previously written scores in that same field.
|
|