KMER Genome Clustering
Introduction:
K-mer Genome Clustering is a powerful technique used to group genomes based on their sequence similarity (without needing traditional sequence alignment).
K-mers are short subsequences of length k extracted from DNA or RNA sequences.
For example, with the sequence ATGG, the 3-mers (k = 3) would be:
- ATG
- TGG
How does it work in general:
-
Each genome is broken down into all possible k-mers.
-
The frequency of each k-mer is counted, creating a k-mer profile for each genome
-
These profiles are compared using distance metrics.
-
Genomes with similar profiles are clustered together using an algorithm.KMER Genomes Clustering in BioloMICS:
Genomes can be stored and analyzed in BioloMICS.
The Download NCBI Genomes tool can be used to retrieve the available genomes and to store them as a .zip file in the database, linked to the associated strains.
Genomes that can be used for the KMER genomes clustering are stored in an Flink field in (zipped or sequence file: Fasta (.fasta, .fas or .fna) or .fastq).
It is also possible add manually genomes to the database.
Then load the zip or sequence file to the Flink field, for instructions see Edit Flink field.
-
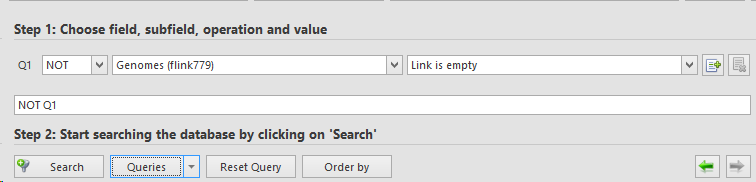
Perform a search to find all the records that have a genome linked.In this example we have an Flink field called Genomes, and we search for all records that have no value in that field.
-
Select the records (with a genome linked) in the grid.
-
To open the KMER Genomes Clustering tool, under Analytics, in the Classification group, click KMER Genomes Clustering.
-
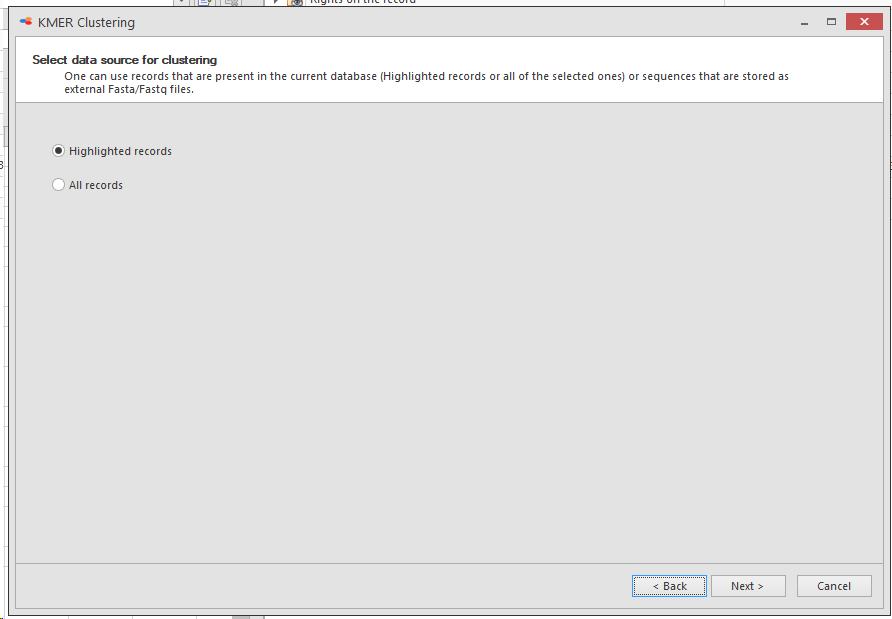
Select the data source for clustering, choose one of the 2 data sources and click on the Next button:
-
Highlighted records: Selected records in the grid only.
-
All records. Note that if a search for specific records was done and the results are 10 records, then 'all' means those 10 records.
-
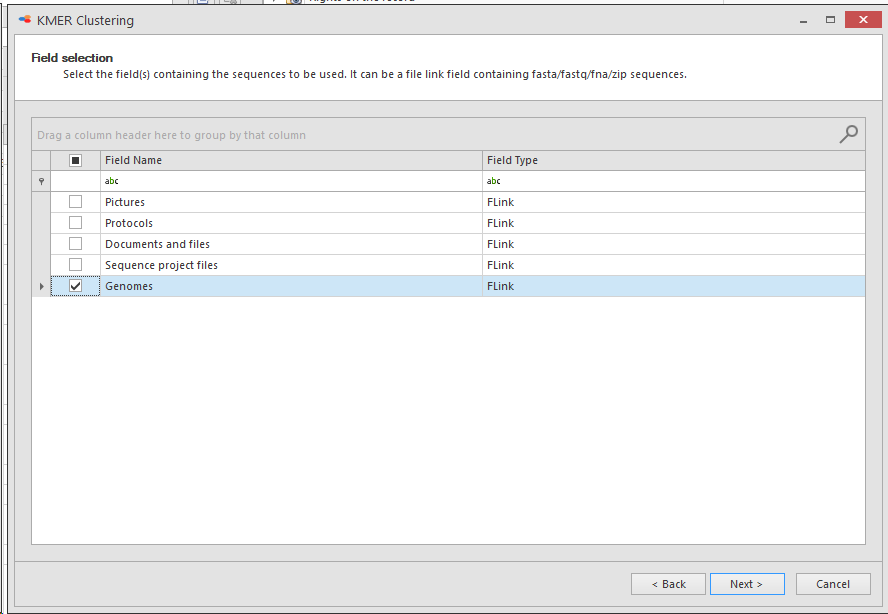
Select the field(s) containing the sequences to be used.
-
Select the following options
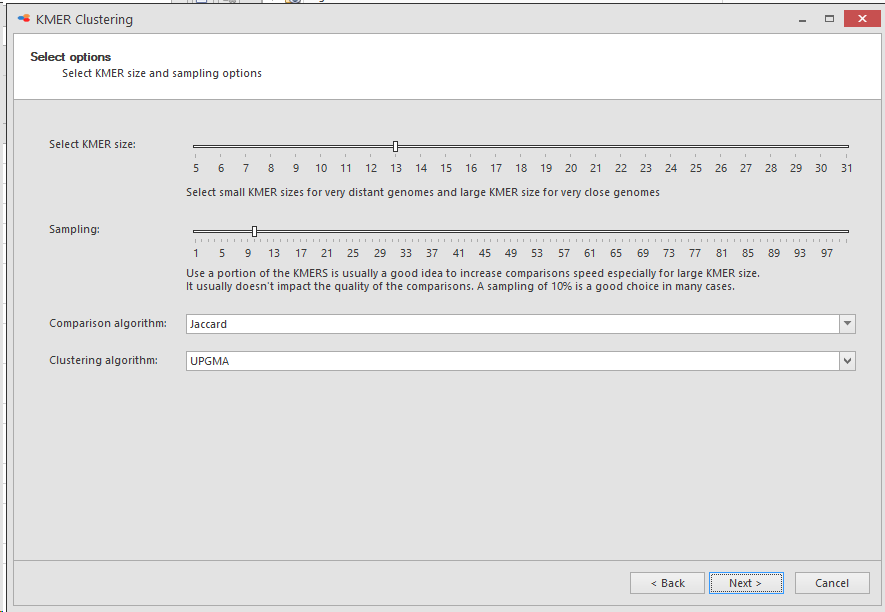
-
Select KMER size: KMER size is the number of base-pairs being used and shifted 1 place each time for the full length of the genome.Select a small KMER size for very distant genomes and a large KMER size of very close genomes. Default is 13.
-
Sampling: Select the portion of the KMERS to be used in this clustering. Default is 10.For speed reasons it is usually good to use a portion of the KMERS especially when using a large KMER size.It usually does not impact the quality of the comparisons.A sampling of 10% is a good choice in many cases.
-
Comparison algorithm: Select the algorithm that will be used for comparing the genomes, the possible algorithms are:
-
Jaccard
-
Min
-
Max
-
Cosine
-
Mash
-
Ani
-
Clustering algorithm: Select the algorithm that will be used to build the distance matrix and the clustering tree, the possible algorithms are:
-
UPGMA
-
WPGMA
-
SINGLE
-
COMPLETE
-
NJ
-
WARD
-
UPGMC
-
LANCE
-
MDS
-
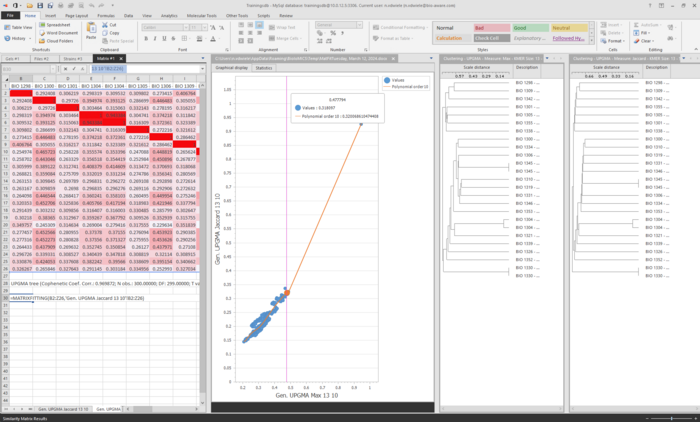
The matrix and the tree will now be displayed
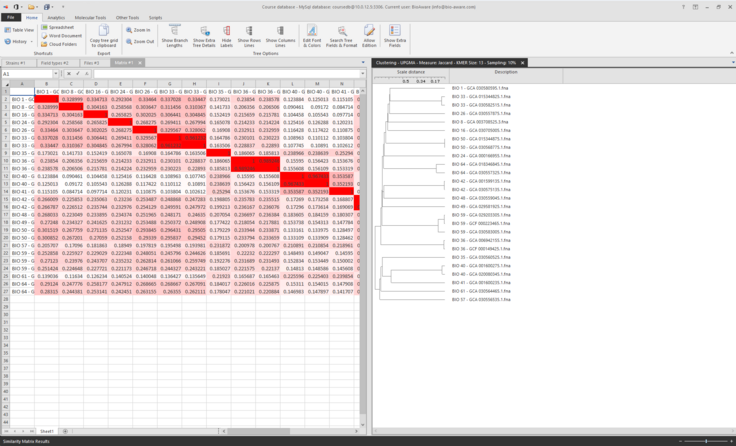
-
Fields can be added besides the tree and colors can also be applied, for more information of the options in the clustering tool, see Agglomerative clustering.
-
Note that the popup window for the KMER clustering is still opened in the background. Changes like KMER size can be made by clicking on the Back button and completing the wizard to the end again. A new matrix and tree will be displayedIt is possible to compare different matrices using the formula Matrixfitting.In one of the 2 matrices, enter in an empty cell "=MATRIXFITTING(select matrix 1,select matrix 2)"A graphic will then be displayed showing the fitting of the 2 matrices used.